Politische Stellungnahmen
Nationale Gesundheitspolitik - Stellungnahmen 2024
04/2024: Stellungnahme zur geplanten Änderung der der Richtlinie für organisierte Krebsfrüherkennungsprogramme (oKFE-RL)
Die Arbeitsgemeinschaft Prävention und integrative Medizin in der Onkologie (PRiO) in der Deutschen Krebsgesellschaft e. V. hat zu einer geplanten Änderung der der Richtlinie für organisierte Krebsfrüherkennungsprogramme (oKFE-RL) Stellung genommen. Die Arbeitsgemeinschaft der DKG spricht sich dafür aus, dass bei den Abgleichen mit Krebsregistern die TNM-Auflage erfasst wird.
03/2024: Gemeinsame Erklärung von Verbänden und Organisationen aus dem Gesundheitswesen
Demokratie und Pluralismus sind Grundvoraussetzungen für ein Leben in Frieden und Freiheit. Sie sind elementar für das Wohlergehen unseres Landes und Fundament für das Zusammenleben und Zusammenwirken in allen Bereichen unseres gesellschaftlichen Miteinanders. Auf dieser Basis steht auch und gerade das Gesundheitswesen in Deutschland. Wir haben mit 200 weiteren Verbänden und Organisationen des Gesundheitsbereichs die gemeinsam Erklärung unterzeichnet.
03/2024: Stellungnahme der Abteilung Experimentelle Krebsforschung (AEK) in der DKG zum Referentenentwurf eines Gesetzes zur Änderung des Tierschutzgesetzes
Die Abteilung Experimentelle Krebsforschung (AEK) der Deutschen Krebsgesellschaft als größte Vereinigung experimentell Krebsforschender in Deutschland unterstützt in allen Punkten die Stellungnahme der Deutschen Forschungsgemeinschaft (DFG) vom 26.02.2024 zum „Entwurf eines Gesetzes zur Änderung des Tierschutzgesetzes und des Tiererzeugnisse-Handels-Verbotsgesetzes“.
02/2024: Stellungnahme zum Referentenentwurf eines Medizinforschungsgesetzes (MFG)
Die Deutsche Krebsgesellschaft hat eine Stellungnahme zum Referentenentwurf eines Medizinforschungsgesetzes abgegeben. In ihrer Stellungnahme begrüßt die Fachgesellschaft bestimmte Maßnahmen zur Verbesserung der Rahmenbedingungen für die medizinische Forschung und stellt gleichzeitig durch den Entwurf vernachlässigte Bereiche heraus.
02/2024: Gemeinsame Stellungnahme zur Bewertung der Lungenkrebsfrüherkennung mittels Niedrigdosis-Computertomographie bei Rauchern
Die Deutsche Krebsgesellschaft hat gemeinsam mit weiteren Fachgesellschaften und Verbänden eine Stellungnahme zur Lungenkrebsfrüherkennung mittels Niedrigdosis-Computertomographie bei Rauchern erarbeitet. In ihrer Stellungnahme sprechen sich die Organisationen unter anderem für die verpflichtende Vorstellung aller abklärungsbedürftigen Befunde in einem interdisziplinären Board in einer auf die Diagnostik und Behandlung von Lungenkrebs spezialisierten Einrichtung wie den von der DKG zertifizierten Lungenkrebszentren aus.
Nationale Gesundheitspolitik - Stellungnahmen 2023
11/2023: Gemeinsame Stellungnahme zum Regierungsentwurf eines Gesetzes zur verbesserten Nutzung von Gesundheitsdaten (Gesundheitsdatennutzungsgesetz – GDNG)
Gemeinsam mit der Arbeitsgemeinschaft Deutscher Tumorzentren und der Deutschen Krebshilfe hat die DKG eine Stellungnahme zum Entwurf des Gesundheitsdatennutzungsgesetzes der Bundesregierung erarbeitet. In ihrer Stellungnahme fordern die Organisationen, das Projekt „Plattform-Lösung zur Datenzusammenführung der Stufe 2 (Plato 2)“ im Gesetzestext zu berücksichtigen. Das Projekt hat zum Ziel, eine Plattform zur Verknüpfung von Daten der Landeskrebsregister sowie weiterer Datenquellen zu konzipieren, wozu die beteiligten Organisationen im Gesetz zur Zusammenführung von Krebsregisterdaten beauftragt worden waren.
Hier geht's zur Stellungnahme.
08/2023: Stellungnahme zum Entwurf eines Gesetzes zur Förderung der Qualität der stationären Versorgung durch Transparenz (Krankenhaustransparenzgesetz)
Die DKG hat Stellung zum Entwurf eines Krankenhaustransparenzgesetzes genommen. Der Entwurf sieht vor, Patient*innen durch ein Transparenzverzeichnis über die Versorgungsqualität in den stationären Einrichtungen zu informieren. Die DKG zeigt die Herausforderungen bei der Umsetzung eines solchen Verzeichnisses im Bereich der Onkologie auf und fordert, die Zertifikate der zertifizierten Zentren im Verzeichnis zu berücksichtigen.
Hier geht's zur Stellungnahme.
08/2023: Stellungnahme zum Beschlussentwurf über die Richtlinie zur Erprobung der Endoskopischen Injektions-Implantation bei irresektablen, lokal fortgeschrittenen Pankreastumoren
Der Gemeinsame Bundesausschuss hat eine Richtlinie zur Erprobung der Endoskopischen Injektions-Implantation von 32P-markierten Mikropartikeln bei irresektablen, lokal fortgeschrittenen Pankreastumoren vorgelegt. Die DKG unterstützt sehr die Durchführung dieser innovativen und für die Patientenversorgung relevanten Studie und merkt in ihrer Stellungnahme die Notwendigkeit der Interdisziplinarität in der Beurteilung potenzieller Studienteilnehmer und in der Beurteilung der ausgewählten Endpunkte an.
Hier finden Sie die Stellungnahme.
08/2023: Stellungnahme zum Entwurf einer Lungenkrebs-Früherkennungs-Verordnung
Die DKG hat eine Stellungnahme zum Entwurf einer Lungenkrebs-Früherkennungs-Verordnung abgegeben. Mit der Verordnung soll die Anwendung der Niedrigdosis-Computertomographie zur Früherkennung von Lungenkrebs bei Rauchern zugelassen werden.
Hier finden Sie die Stellungnahme.
08/2023: Stellungnahme zum Referentenentwurf eines Gesetzes zur verbesserten Nutzung von Gesundheitsdaten (Gesundheitsdatennutzungsgesetz – GDNG)
Unter Mitwirkung ihrer Arbeitsgemeinschaft Internistische Onkologie (AIO) hat die Deutsche Krebsgesellschaft e. V. (DKG) eine Stellungnahme zum Referentenentwurf des Gesundheitsdatennutzungsgesetzes (GDNG) erarbeitet. In ihrer Stellungnahme fordert die DKG, die Verknüpfung von Gesundheitsdaten zu Forschungszwecken breiter als im Entwurf vorgesehen zu ermöglichen und bislang bestehende Möglichkeiten der Forschung nicht einzuschränken. Zudem weist die DKG darauf hin, die im Gesetz zur Zusammenführung von Krebsregisterdaten beauftragte Erstellung einer Plattform zur anlassbezogenen Zusammenführung und Analyse von Daten der Krebsregister sowie weiterer Daten (Plato2) im GDNG zu berücksichtigen.
Hier geht's zur Stellungnahme.
08/2023: Stellungnahme zum Entwurf eines Gesetzes zur Beschleunigung der Digitalisierung des Gesundheitswesens (Digital-Gesetz – DigiG)
Die Deutsche Krebsgesellschaft e. V. (DKG) hat eine Stellungnahme zum Referentenentwurf des Digitalgesetzes erarbeitet. In ihrer Stellungnahme geht die DKG auf die vorgesehenen Änderungen in Bezug auf den Innovationsfonds ein und regt an, weitere Maßnahmen zur Verbesserung der Wirksamkeit des Innovationsfonds in das Gesetz aufzunehmen.
Hier geht's zur Stellungnahme.
04/2023: Gemeinsames Positionspapier der onkologisch tätigen Fachgesellschaften der AWMF Ad hoc Kommission Versorgungsstrukturen
Zusammen mit anderen Fachgesellschaften habe wir zur Krankenhausreform Stellung genommen: in einem gemeinsamen Positionspapier der onkologisch tätigen Fachgesellschaften der AWMF Ad hoc Kommission Versorgungsstrukturen zu der „Dritten Stellungnahme und Empfehlung der Regierungskommission für eine moderne und bedarfsgerechte Krankenhausversorgung mit grundlegender Reform der Krankenhausvergütung“.
Hier geht's zum Positionspapier.
04/2023: Stellungnahme der PSO zur Überarbeitung des Disease-Management-Programms Brustkrebs
Die Arbeitsgemeinschaft für Psychoonkologie in der Deutschen Krebsgesellschaft e. V. (PSO) hat eine Stellungnahme zur Überarbeitung des Disease-Management-Programms (DMP) Brustkrebs abgegeben. In ihrer Stellungnahme verwies die Arbeitsgemeinschaft darauf das Screening auf Belastung (Distress) sowie das Versorgungsangebot der Krebsberatungsstellen im DMP zu berücksichtigen.
04/2023: Gemeinsame Stellungnahme zur Ernährungsmedizin im Krankenhaus
Gemeinsam mit 22 weiteren Fachgesellschaften hat sich die Deutsche Krebsgesellschaft e. V. zur Ernährungsmedizin im Krankenhaus positioniert. In ihrer Stellungnahme fordern die Fachgesellschaften ein systematisches Screening auf Mangelernährung sowie im Zuge der Krankenhausreform die Einführung einer im Qualitätsmanagement verankerten Ernährungskompetenz als Teil der Mindeststrukturvoraussetzungen in allen drei Versorgungsstufen. Zugleich soll dieses Präventions- und Therapiekonzept vom DRG-System abgebildet und entsprechend vergütet werden.
03/2023: Stellungnahme des Ausschusses Krebsberatung der Landeskrebsgesellschaften zur Förderung nach § 65e SGB V
Die Fördergrundsätze des GKV-Spitzenverbandes bilden die Grundlage für die finanzielle Förderung ambulanter Krebsberatungsstellen (KBS) durch den GKV-Spitzenverband und die Private Krankenversicherung. Sie sind abgeleitet aus dem Gesetzestext des § 65e SGB V auf Basis der zugrundeliegenden „Empfehlungen für das Leistungsspektrum, die Qualitätskriterien und für Finanzierungsmodelle ambulanter psychosozialer Krebsberatungsstellen“. Nach Abschluss der ersten Förderperiode durch den GKV-Spitzenverband (2020–2022) nimmt der Ausschuss Krebsberatung, ein Gremium der Landeskrebsgesellschaften, aus der Praxis dazu Stellung. Aus Sicht des Ausschusses ergeben sich auf Grundlage der derzeitigen Förderpraxis weiterhin Herausforderungen für die nachhaltige Finanzierung von KBS.
01/2023: Stellungnahme der AG OPH zum Austausch von Biosimilars
Die Arbeitsgemeinschaft Onkologische Pharmazie in der Deutschen Krebsgesellschaft e. V. (AG OPH) hat eine Stellungnahme zu einer geplanten Änderung der Arzneimittelrichtlinie erarbeitet. Durch den neuen §40b sollen Apotheken bei der Abgabe verordneter biotechnologisch hergestellter biologischer Arzneimittel an Versicherte unter bestimmten Voraussetzungen zur Ersetzung durch ein preisgünstiges Arzneimittel verpflichtet werden. Die AG OPH verweist darauf, dass der Gemeinsame Bundesausschuss bei der Umsetzung des gesetzlichen Auftrags behutsam vorgesehen solle.
01/2023: Positionspapier zu Arzneimittelengpässen in der Behandlung von Krebspatient*innen
Die Deutsche Krebsgesellschaft e. V. hat sich gemeinsam mit der Deutschen Gesellschaft für Hämatologie und medizinische Onkologie (DGHO) e. V. und der Deutschen Gesellschaft für Gynäkologie und Geburtshilfe (DGGG) e. V. zu Arzneimittelengpässen in der Behandlung von Krebspatient*innen positioniert. In einem Positionspapier haben die Fachgesellschaften verschiedene Forderungen formuliert, bspw. die frühzeitige Information über drohende Lieferengpässe seitens der pharmazeutischen Unternehmen.
Nationale Gesundheitspolitik - Stellungnahmen 2022
11/2022: Stellungnahme zur Entwicklung von Follow-up-Indikatoren durch das IQTIG
In einer Stellungnahme hat die Deutsche Krebsgesellschaft e. V. sich zum Vorbericht des IQTIG „Methodik für die Entwicklung von Follow-up-Indikatoren und die Beurteilung ihrer Zuschreibbarkeit“ positioniert. Aus Sicht der Deutschen Krebsgesellschaft ist die Onkologie für die Anwendung von Follow-up-Indikatoren nicht geeignet.
08/2022: Stellungnahme zur Brustkrebsfrüherkennung mittels Röntgenmammographie bei Frauen ab 70 Jahren
Das Bundesamt für Strahlenschutz (BfS) hat bewertet, welchen Nutzen und welche Risiken die Fortführung des Mammographie Screening-Programms bei Frauen über 70 Jahren hätte. Die Deutsche Krebsgesellschaft (DKG) e. V. und die Arbeitsgemeinschaft Internistische Onkologie (AIO) in der DKG e. V. befürworten klar die Brustkrebsfrüherkennung mittels Mammographie in zweijährigen Intervallen auch für Frauen im Alter von 70 bis 74 Jahren.
08/2022: Stellungnahme der AG GePoMAx zum Wert von Kombinationstherapien
Die Arbeitsgemeinschaft Gesundheitspolitik und Market Access der Sektion C der Deutschen Krebsgesellschaft e. V. (AG GePoMAx) hat eine Stellungnahme zum Wert von Kombinationstherapien erarbeitet. Hintergrund der Stellungnahme ist die im Kabinettsentwurf des GKV-Finanzstabilisierungsgesetzes vorgesehene Einführung eines Kombinationsabschlages.
07/2022: Stellungnahme zu Kriterien des IQTIG zur Bewertung von Zertifikaten und Qualitätssiegeln
Die Deutsche Krebsgesellschaft e. V. hat zum Zwischenbericht des IQTIG „Entwicklung von Kriterien zur Bewertung von Zertifikaten und Qualitätssiegeln gemäß § 137a Absatz 3 Satz 2 Nummer 7 SGB V“ Stellung genommen. Die Deutsche Krebsgesellschaft begrüßt die Entwicklung der Kriterien, um Patient*innen eine Grundlage zu bieten, Zertifikate und Qualitätssiegel systematisch miteinander zu vergleichen. Gleichzeitig stellt sie in ihrer Stellungnahme die hohen Anforderungen an Zertifikate heraus.
04/2022: Empfehlungen zum Umgang mit der elektronischen Zigarette (E-Zigarette)
Unter Federführung der Deutschen Gesellschaft für Pneumologie hat die Deutsche Krebsgesellschaft gemeinsam mit 13 weiteren Fachgesellschaften und Organisationen ein Positionspapier mit Empfehlungen zum Umgang mit der elektronischen Zigarette (E-Zigarette) erarbeitet.
02/22: Gemeinsame Stellungnahme zu den Entwürfen der Gesundheitsinformationen zum Themenpaket „Rauchen“ des IQWiG
Die Arbeitsgemeinschaft Prävention und integrative Onkologie (PRiO) der Deutschen Krebsgesellschaft und die Deutsche Krebsgesellschaft e. V. haben eine gemeinsame Stellungnahme zu den Entwürfen für eine Aktualisierung der Informationen zum Themenpaket „Rauchen“ auf gesundheitsinformation.de durch das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) abgegeben.
02/2022: Stellungnahme der AG Gynäkologische Onkologie und weiterer Fachgesellschaften zum Lieferengpass Tamoxifen
Anbei finden Sie die gemeinsame Stellungnahme der Arbeitsgemeinschaft Gynäkologische Onkologie (AGO) in der Deutschen Krebsgesellschaft und weiterer Fachgesellschaften zum Lieferengpass von Tamoxifen in der Behandlung von Brustkrebspatientinnen.
Nationale Gesundheitspolitik - Stellungnahmen 2021
11/2021: Stellungnahme der DGHO in Zusammenarbeit mit acht weiteren Organisationen zur Einführung des E-Rezepts.
Die Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) und acht weitere Organisationen nehmen Stellung zur Einführung des E-Rezepts am 01.01.2022. Aufgrund fehlender Eignung bisheriger Konzepte für komplexe onkologische Therapien werden die Verschiebung der Umstellung und eine Pilotphase vorgeschlagen.
08/2021: Gemeinsame Stellungnahme zu den Fördergrundsätzen des GKV-Spitzenverbandes zu ambulanten Krebsberatungsstellen
Die Bundesarbeitsgemeinschaft für Krebsberatungsstellen, die Deutsche Krebsgesellschaft, die Landeskrebsgesellschaften in der DKG und die Stiftung Deutsche Krebshilfe haben zu den Fördergrundsätzen des GKV-Spitzenverbandes für ambulante Krebsberatungsstellen gemäß § 65e SGB V Stellung genommen.
05/2021: Stellungnahme zum Vorbericht des IQTIG "Entwicklung eines Qualitätssicherungsverfahrens ‚Lokal begrenztes Prostatakarzinom‘"
Die Deutsche Krebsgesellschaft hat zum Vorbericht des Instituts für Qualitätssicherung und Transparenz im Gesundheitswesen (IQTIG) „Entwicklung eines Qualitätssicherungsverfahrens ‚Lokal begrenztes Prostatakarzinom‘“ Stellung genommen.
04/2021: Stellungnahme zum Entwurf des Gesundheitsversorgungsweiterentwicklungsgesetzes
Die Deutschen Krebsgesellschaft hat zum Änderungsantrag der Fraktionen CDU/CSU und SPD zum Entwurf des Gesetzes zur Weiterentwicklung der Gesundheitsversorgung (Gesundheitsversorgungsweiterentwicklungsgesetz – GVWG) Stellung genommen: Die Stellungnahme bezieht sich auf
§ 64 d SGB V zum „Modellvorhaben zur umfassenden Diagnostik und Therapiefindung mittels Genomsequenzierung bei seltenen und onkologischen Erkrankungen“. Der Paragraph bietet eine bisher nicht im SGB V vorhandene Möglichkeit, Leistungserbringer nach Qualitätskriterien selektiv auszuwählen.
02/2021: Stellungnahme zum Entwurf des Gesundheitsversorgungsweiterentwicklungsgesetzes: ambulante Krebsberatung
Mit dem Gesetzentwurf zur Sicherstellung einer nachhaltig gesicherten Finanzierung ambulanter Krebsberatungsstellen wird die bereits in § 65e SGB V vorgesehen Finanzierung aufgestockt. Die Krebsberatung soll die psychologischen und sozialen Aspekte einer solchen Beratung umfassen. Die Deutsche Krebsgesellschaft begrüßt dieses Vorhaben ausdrücklich.
01/2021: Stellungnahme zum Entwurf eines Gesetzes zur Zusammenführung von Krebsregisterdaten
Die Mitinitiatoren des Nationalen Krebsplanes - Deutschen Krebsgesellschaft, Deutsche Krebshilfe und Arbeitsgemeinschaft Deutscher Tumorzentren - haben zum Entwurf eines Gesetzes zur Zusammenführung von Krebsregisterdaten vom 10.12.2020 Stellung genommen: Das Gesetz hat eine hohe Bedeutung, weil es eine optimale Nutzung der gesammelten Daten für die Verbesserung der Versorgung und die Fortentwicklung der onkologischen Medizin ermöglicht.
Nationale Gesundheitspolitik - Stellungnahmen 2020
11/2020: Stellungnahme zum Entwurf eines Gesetzes zur Weiterentwicklung der Gesundheitsversorgung (Gesundheitsversorgungsweiterentwicklungsgesetz – GVWG)
Mit dem Referentenentwurf zur Weiterentwicklung der Gesundheitsversorgung (GVWG) legt das Bundesministerium für Gesundheit ein umfangreiches Regelwerk vor, das zahlreiche Adjustierungen im Gesundheitswesen vornimmt. Mit den Weiterentwicklungen der Vorgaben, insbesondere zu Qualitätsverträgen, Mindestmengen und Transparenz in der Versorgung, stellt der Referentenentwurf die Messung und den Nachweis der Qualität der Betreuung aller Patient*innen in den Vordergrund. In ihrer Stellungnahme konzentriert sich die Deutsche Krebsgesellschaft auf die genannten Bereiche, die die Versorgung onkologischer Patient*innen und besonders die Versorgung innerhalb Onkologischer Zentren betreffen.
11/2020: Positionspapier "Verfahren der Ethikberatung von multizentrischen Nicht-AMG/MPG-Studien vereinheitlichen"
Die aktuelle Praxis bei der Ethikberatung von multizentrischen, nichtinterventionellen Studien ist zeitaufwändig, teuer und stellt Forschungsprojekte nichtkommerzieller Förderer vor enorme finanzielle und zeitliche Herausforderungen. Vereinheitlichte Verfahren werden bereits durch nationale sowie europäische Vorgaben begünstigt und haben sich im Ausland bewährt. Die Deutsche Krebsgesellschaft fordert deshalb ein vereinheitlichtes, zentral koordiniertes Verfahren der Ethikberatung von multizentrischen Nicht-AMG/MPG-Studien, das klar definierte Anforderungen, Zeitrahmen, Kosten und Bearbeitungsstrukturen schafft.
09/2020: Stellungnahme der Deutschen Krebsgesellschaft zum Antrag der Fraktion der FDP im Thüringer Landtag
Die Deutsche Krebsgesellschaft hat zum Antrag der Fraktion der FDP im Thüringer Landtag "Dem Krebs den Kampf ansagen – wirksame Therapien fördern, Neuerkrankungen reduzieren, Patientinnen und Patienten bestmöglich unterstützen" (Drucksache 7/682 vom 29.04.2020) am 29.09.2020 Stellung genommen.
09/2020: Stellungnahme an den Gemeinsamen Bundesausschuss zu den Zentrums-Regelungen
Stellungnahme der Deutschen Krebsgesellschaft zur Änderung der Regelungen zur Konkretisierung der besonderen Aufgaben von Zentren und Schwerpunkten gemäß § 136c Absatz 5 SGB V (Zentrums-Regelungen)
08/2020: Gemeinsame Stellungnahme zum MTA-Gesetz
Gemeinsame Stellungnahme von neun Fachgesellschaften und Verbänden, einschließlich der Deutschen Krebsgesellschaft - vertreten durch die Arbeitsgemeinschaft Bildgebung in der Onkologie (ABO) der DKG, zum Entwurf eines Gesetzes zur Reform der technischen Assistenzberufe in der Medizin und zur Änderung weiterer Gesetze.
07/2020: Krebs und Armut - Positionspapier der Deutschen Krebshilfe und des Hauses der Krebs-Selbsthilfe - Bundesverband e. V.
Eine Krebserkrankung beeinflusst die wirtschaftliche Situation der meisten betroffenen Menschen. Bezug nehmend auf das Bundesteilhabegesetz (BTHG) von 2016 sowie den 2. Teilhabebericht des Bundesministeriums für Arbeit und Soziales aus dem Jahr 2017 beschreibt dieses Positionspapier die aktuelle Situation im Rahmen einer Bestandsaufnahme, benennt konkrete Probleme und Defizite und formuliert darauf basierende Forderungen.
01/2020: Empfehlungen für das Leistungsspektrum, die Qualitätskriterien und für Finanzierungsmodelle ambulanter psychosozialer Krebsberatungsstellen
Im Rahmen des Nationalen Krebsplans (NKP) hat die vom BMG moderierte Arbeitsgruppe „Qualitätssicherung und Finanzierungsmodelle für Krebsberatungsstellen“ (AG KBS) gemeinsam mit maßgeblichen Expert*innen ein Papier "Empfehlungen für das Leistungsspektrum, die Qualitätskriterien und für Finanzierungsmodelle ambulanter psychosozialer Krebsberatungsstellen" erarbeitet und im Januar 2020 veröffentlicht.
Nationale Gesundheitspolitik - Stellungnahmen 2019
11/2019: Stellungnahme zur Änderung der Dokumentationsvorgaben nach der Richtlinie für organisierte Krebsfrüherkennungsprogramme
Die DKG hat zur Änderung der Dokumentationsvorgaben nach der Richtlinie für organisierte Krebsfrüherkennungsprogramme Stellung genommen: Aufhebung des Beschlusses zum Wechsel der Dokumentationsvorgaben zum Darmkrebsscreening und Aussetzung der Dokumentationsvorgaben zum Zervixkarzinomscreening.
10/2019: Stellungnahme zum Beschlussentwurf des Gemeinsamen Bundesausschusses über Regelungen zur Konkretisierung der besonderen Aufgaben von Zentren und Schwerpunkten
Im Krankenhausentgeltgesetz hat der Gesetzgeber die Teilfinanzierung von Zentren über Zentrumszuschläge festgelegt (§ 136c, Abs. 5, SGB V). Der Gemeinsame Bundesausschuss ist damit beauftragt, Regelungen zur Konkretisierung der besonderen Aufgaben von Zentren und Schwerpunkten zu definieren. Im August hat er einen ersten Beschlussentwurf vorgelegt. Gemeinsam mit 19 Fachgesellschaften und einschließlich 14 DKG-Arbeitsgemeinschaften, die im Zertifizierungssystem der Deutschen Krebsgesellschaft aktiv sind, haben wir dazu Stellung genommen.
08/2019: Forderungen zur Einführung hochpreisiger neuer Behandlungsverfahren am Beispiel der CAR-T-Zelltherapie
Ein weiterer Schritt für eine Wissen generierende onkologische Versorgung: Ein Bündnis aus Ersatzkassen, der Deutschen Hochschulmedizin, Deutscher Krebsgesellschaft und Deutscher Gesellschaft für Hämatologie und Medizinische Onkologie fordern die kontrollierte Einführung von CAR-T-Zellen und anderen hochpreisigen Arzneimitteln in Innovationszentren mit begleitender Qualitätssicherung. Ein abgestimmter Forderungskatalog wurde im Rahmen einer gemeinsamen Pressekonferenz im September 2019 vorgestellt. DKG-Generalsekretär Dr. Johannes Bruns forderte darin gleichzeitig den raschen Zugang für alle Patient*innen, die von solchen Therapien profitieren, und eine qualitätsgesicherte Anwendung sowie die Sammlung und Evaluation aller Behandlungsdaten zur raschen Auswertung. Denn ohne diese Daten ist die Entscheidung über eine Aufnahme neuer Therapien in die Regelfinanzierung kaum möglich.
06/2019: Stellungnahme der DKG zum Digitale Versorgung Gesetz (DVG)
Das Bundesministerium für Gesundheit legt mit dem Digitale Versorgung Gesetz (DVG) ein umfangreiches Regelungswerk zur Implementierung digitaler Prozesse und Standards im Gesundheitswesen vor. Grundsätzlich begrüßt die Deutsche Krebsgesellschaft diese Aktivitäten, nimmt jedoch nicht im Detail dazu Stellung. Darüber hinaus regelt der Entwurf jedoch auch neue Maßnahmen für die Fortsetzung des Innovationsfonds. Das Bundesministerium sieht in seinem Referentenentwurf vor, darüber auch die Finanzierung von Leitlinienentwicklungen zu regeln. Die vorliegende Stellungnahme der Deutschen Krebsgesellschaft konzentriert sich ausschließlich auf diesen Punkt und kommentiert keine weiteren Regelungsvorschläge des DVG.
2 Positionspapiere vom Aktionsbündnis Nichtrauchen e.V. (ABNR) zu Tabakerhitzern und zu E-Zigaretten
Das Aktionsbündnis Nichtrauchen e. V. (ABNR), dessen Mitglied die Deutsche Krebsgesellschaft ist, hat zwei Positionspapiere veröffentlicht - zum einen zu Tabakerhitzern, zum anderen zu E-Zigaretten.
01/2019: Positionspapier "Qualitätsgesicherte Molekulardiagnostik in der Onkologie: zielgerichtet – integriert"
Diagnostik und Therapie maligner Erkrankungen unterliegen einem tiefgreifenden Wandel. Krebs spaltet sich heute in einer Vielzahl von verschiedenen Erkrankungen mit jeweils eigenen Merkmalen auf. Therapeutische und zunehmend zielgerichtete Maßnahmen basieren auf einer differenzierten Diagnostik. Dabei spielt die molekulare Diagnostik eine zunehmend wichtige Rolle. Angesichts der Vielzahl der diagnostischen Möglichkeiten haben betroffene wissenschaftliche Fachgesellschaften ihre Position zum Einsatz der Molekulardiagnostik in der Versorgung von Krebspatienten definiert und am 15. Januar 2019 in Berlin vorgestellt.
Nationale Gesundheitspolitik - Stellungnahmen 2018
12/2018: Stellungnahme zum Entwurf für ein "Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV)"
Am 20.11.2018 veröffentlichte das Bundesministerium für Gesundheit einen Entwurf für ein „Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV)“. Mit dem Gesetz reagiert der Gesetzgeber auf verschiedene sicherheitsrelevante Ereignisse, die in den vergangenen Monaten stattfanden und in vielen Fällen auch die Onkologie betrafen. Das Ministerium nutzt aber auch die Gelegenheit einige weitere Punkte zu regeln, unter anderem die anwendungsbezogene Datenerhebung bei bestimmten neu zugelassenen Arzneimitteln. Zu diesem Punkt nahm die Deutsche Krebsgesellschaft am 14.12.2018 Stellung.
11/2018: Stellungnahme zum Referentenentwurf der AIS-Rechtsverordnung
Das Arzneimittelversorgungsstärkungsgesetz, das bereits seit 2017 in Kraft ist, sieht vor, dass die Beschlüssen des G-BA zur frühen Nutzenbewertung den niedergelassenen Ärzten über die Praxissoftware (Arztinformationssystem) zugänglich gemacht werden. Der entsprechende Entwurf einer Rechtsverordnung vom 15. Oktober 2018 des Bundesministeriums für Gesundheit liegt nun vor. Die Deutsche Krebsgesellschaft nimmt gemeinsam mit der Ad-hoc-Kommission "Nutzenbewertung von Arzneimitteln" der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) Stellung zum Referentenentwurf.
11/2018: Stellungnahme des Bereichs Zertifizierung in der Deutschen Krebsgesellschaft zum Zweiten Gesetz zur Änderung des Thüringer Krankenhausgesetzes
Mit dem Krankenhausstrukturgesetz, das zum 1. Januar 2016 in Kraft getreten ist, sollten planungsrelevante Qualitätsindikatoren Bestandteil der Krankenhausplanung werden. Das Land Thüringen hat in einem Gesetzentwurf für die Landesebene die automatische Übernahme dieser Qualitätsindikatoren ausgeschlossen. Sie befürchtet durch drohende Abschläge wirtschaftliche Probleme für einzelne Krankenhäuser. Der Bereich Zertifizierung in der Deutschen Krebsgesellschaft hat zum Gesetzesentwurf Stellung bezogen.
Endbericht: Nutzen, Mehraufwand und Finanzierung von Onkologischen Spitzenzentren, Onkologischen Zentren und Organkrebszentren
Die Stiftung Deutsche Krebshilfe und die Deutsche Krebsgesellschaft haben die Prognos AG mit der Erstellung eines Gutachtens zur Ermittlung des zentrumsspezifischen Mehraufwandes an Onkologischen Spitzenzentren, Onkologischen Zentren und Organkrebszentren sowie der daraus resultierenden Mehrkosten beauftragt. Das Gutachten steht auf der Webseite der Stiftung Deutschen Krebshilfe unter folgendem Link zur Verfügung:
03/2018: Stellungnahme zum Konzept des IQWiG für ein Nationales Gesundheitsportal
Das vom IQWiG vorgelegte Konzept zum Nationalen Gesundheitsportal stellt einen ersten Entwurf dar, der aus Sicht der DKG geeignet ist, die Diskussion verschiedener Anbieter von Gesundheitsinformationen über eine Zusammenarbeit anzuregen. Es fehlt aus DKG-Sicht bislang eine umfassende kritische Analyse, die neben den möglichen Chancen auch alle potenziellen Risiken und Maßnahmen, wie mit diesen umgegangen werden kann, benennt.
Nationale Gesundheitspolitik - Stellungnahmen 2017
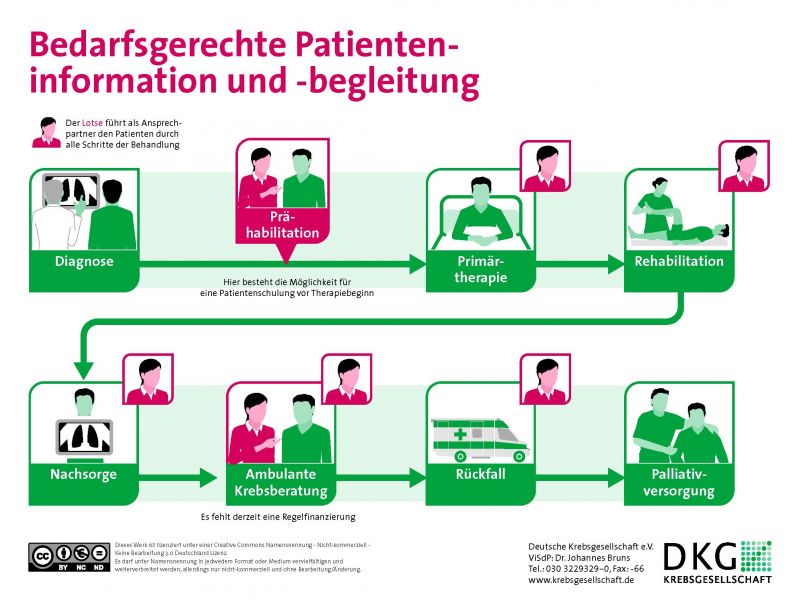
03/2017: Positionspapier "Wissen generierende onkologische Versorgung"
Auf einer Pressekonferenz am 6. März 2017 wurde ein Positionspapier zur Zukunft der onkologischen Versorgung vorgestellt. Die darin formulierten Konzepte der wissensgenerierenden onkologischen Versorgung und einer verbesserten Patientenbegleitung wurden in den vergangenen 1,5 Jahren gemeinsam mit Bundestagsabgeordneten, Vertretern von Krankenkassen, der ambulant und stationär tätigen Ärzteschaft, Wissenschaft und Patientenorganisationen entwickelt. Das Positionspapier präsentiert konkrete Ansätze, die über alle Interessengegensätze hinaus einen tragfähigen Konsens darstellen.
{kind=link}
{kind=link}
03/2017: Grundsatzpapier des UV-Schutz-Bündnisses
20 Institutionen des UV-Schutz-Bündnisses - darunter die Deutsche Krebsgesellschaft - haben das Grundsatzpapier "Vorbeugung gesundheitlicher Schäden durch die Sonne - Verhältnisprävention in der Stadt und auf dem Land" verfasst und veröffentlicht. Es dient dem Ziel, im Freien, in Außenanlagen öffentlicher Einrichtungen sowie in den unterschiedlichen Lebenswelten der Bevölkerung verhältnispräventive Maßnahmen zum Schutz vor übermäßiger UV-Belastung und vor weiteren durch den Klimawandel zunehmenden gesundheitsschädigenden Belastungen der Sonne (z. B. Hitzebelastung) flächendeckend zu etablieren.
01/2017: Zielepapier zum Ziel 11b des Nationalen Krebsplans
Ein Expertengremium hat zum Ziel 11b „Qualitätsgesicherte Beratungs- und Hilfsangebote für Krebspatient(inn)en und ihre Angehörigen“ im Handlungsfeld 4 des Nationalen Krebsplans ein Zielepapier konsentiert und im Januar 2017 veröffentlicht. In dem Papier werden eine Ist- und Soll-Beschreibung der Inhalte von Krebsberatung und der Bereitstellung von Hilfsangeboten in Bezug auf deren Qualität gegeben. Zusätzlich werden Handlungsempfehlungen für eine Qualitätsentwicklung dargestellt und der Forschungsbedarf aufgezeigt. Folgende Schwerpunkt werden dabei gesetzt:
- Sicherung der Qualität und Seriosität der verfügbaren Beratungs- und Hilfsangebote
- Bessere Vernetzung und Vereinheitlichung der vorhandenen Angebote für Krebspatienten und ihre Angehörigen
- Schaffung niederschwelliger zielgruppengerechter Angebote zur besseren Steuerung/Lotsung des Krebspatienten/der Krebspatientin durch das Gesundheitssystem